*Corresponding Author:
Shahin Asadi,
Division of Medical Genetics and Molecular Pathology Research, Harvard University, Boston Children’s Hospital, Iran
Tel: +98 9379923364
E-mail: shahin.asadi1985@gmail.com
Abstract
Fibrodysplasia Ossificans Progressiva (FOP) is a severely disabling heritable disorder of connective tissue characterized by congenital malformations of the great toes and progressive heterotopic ossification that forms qualitatively normal bone in characteristic extraskeletal sites. Classic FOP is caused by a recurrent activating mutation (617G>A; R206H) in the gene ACVR1 (ALK2) encoding Activin A receptor type I/Activin-like kinase 2, a bone morphogenetic protein (BMP) type I receptor. Atypical FOP patients also have heterozygous ACVR1 missense mutations in conserved amino acids.
Keywords
ACVR1; BMPR1A; BMPR1B; BMPR2; BMP4; Genetics mutations, Stone man syndrome
Overview of Stone Man Syndrome
Stone Man Syndrome is a very rare genetic disorder known as progressive bone fibrodysplasia (FOP) or severe progressive bone malformation. Progressive bone fibrodysplasia is a genetic disorder of connective tissue that is associated with abnormal bone development in different areas of the body and usually involves ligaments, tendons, and skeletal muscles. Other skeletal abnormalities of the spine, neck and ribs and abnormal bone growth in the affected areas such as the neck, shoulder, elbows, knee joint, wrist, ankle and jaw are also evident in human stone syndrome (Figure 1).
Bone soft tissue swelling from progressive bone fibrodysplasia (FOP) usually begins during early childhood and develops through- out the life of the affected person [1].

Figure 1: Photographs of the oldest man with stone man syndrome, note the abnor- mal development of bones in the front trunk, head, legs and hands.
Symptoms and Symptoms of Stone Man Syndrome
All people with lithospheric syndrome have common anomalies in the big toes and about 50% of the sums of anomalies in the thumb. These changes are present in the skeletal structure of patients at birth and are the first clinical signs of this disorder. Other abnormalities of the toes are the big toe rotation inward or toward the other toes and the small toes adhere to each other and the constant curvature of the fifth toe. Other congenital signs of stone man syndrome include prox- imal internal tibia, upper spine abnormalities, short abnormal broad neck, and short abnormal thigh bone that extend from the knee to the hip (Figure 2) [2].
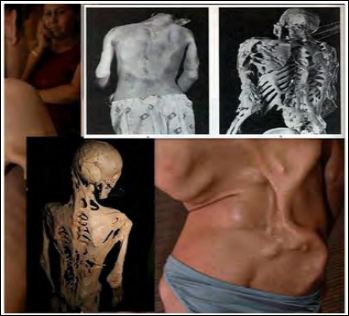
Figure 2: Human and skeletal images found with stone man syndrome (FOP), ob- serve abnormal bone abnormalities in the spine.
Abnormal bone development in stone man syndrome, it occurs spontaneously, but usually begins with soft tissue damage caused by viral disease, abnormal bone development. Abnormal bone growth is commonly seen in tendons, ligaments, skeletal muscle tissue, connec- tive tissue such as fascia and muscle fibers. In some cases, pain and stiffness occur in these areas. Also, in some cases, a low grade fever may promise bone swelling. The neck, back, chest, arms and legs are usually the first affected areas. The condition may eventually affect the hip, ankle, wrist, elbow, shoulder, jaw, and abdominal wall. It is worth noting that in some people with stone man syndrome, bone growth may be rapid and in others it may be gradual (Figure 3) [3].

Figure 3: Picture of man’s hands with stone man syndrome (FOP).
Even among twins, disease progression can vary greatly. Chronic swelling in different parts of the body is a common physical feature of people with stone man syndrome (FOP). Swelling may be associated with abnormal bone, which is characteristic of FOP, or stone man syn- drome. When new bone swelling develops, it may press the bone layer into the lymphatic vessels, which can block blood flow or tissue fluid and in some cases can be life-threatening. In addition, bone swelling may also be caused by a lack of pumping blood into the hard muscles, which causes blood and tissue fluid to accumulate in one of the or- gans, such as the arms or legs [4].
Abnormal bone growth can ultimately lead to joint stiffness and inactivity and movement of the affected person. If the jaw is also involved in stone man syndrome, people with nutrition, especially swallowing and talking, will have trouble. In addition, abnormal bone growth may result in gradual deformity of the spine (scoliosis) or cur- vature of the spine (kyphosis) (Figure 4). In some cases, bone that develops in abnormal areas may cause bone fractures. As the disease progresses, people with FOP will experience limitations in walking, imbalances in the body, and imbalances in sitting [5].
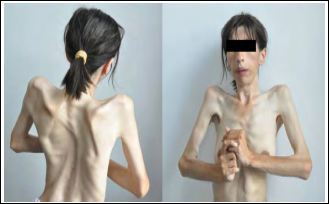
Figure 4: heraldsopenaccess.us/webmail(abnormal curvature of the spine).
Stone man syndrome (FOP) may eventually cause complete im- mobilization of patients. People with this syndrome may feel pain and stiffness in the affected areas. Due to the abnormal growth of the bones in these individuals, their bones, including the bones of the pelvis and calf, may be fractured while walking. They may also show increased respiratory sensitivity and congestion. In some cases of peo- ple with FOP, people may also have hair loss, especially scalp hair. It is worth noting that hearing impairment is found in 50% of people with LBD (Figure 5) [6].
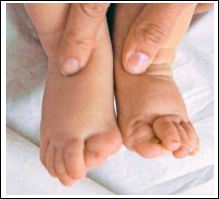
Figure 5: Image of baby legs with stone man syndrome, beware of curved toes.
The Etiology of Stone Man Syndrome
Most cases of stone man syndrome (FOP) occur sporadically. Human stone syndrome follows the dominant autosomal inherited pattern with full gene penetration. Stone man syndrome (FOP) is caused by a genetic mutation in the ACVR1 gene located on the long arm of chromosome 2 as 2q24.1. (Figure 6) It plays a role in the BMP receptor signaling pathway that determines the fate of stem cells. BMPR receptors in humans include BMPR1A, BMPR1B, BMPR2, and BMP4, which play prominent roles in the FOP signaling pathway of only BMPR1A and BMP4 receptors. The ACVR1 gene encodes the bone marrow receptor protein (BMP) that encodes the BMPR1A receptor [7].

Figure 6: Schematic overview of chromosome 2 where the ACVR1 gene is located in the long arm of chromosome 2q24.1.
The BMPR1A gene is located on the long arm of chromosome 10 as 10q23.2. (Figure 7) Mutation in the ACVR1 gene replaces arginine amino acids instead of histidine at the codon 206 position of the ACVR1 protein. This amino acid substitution causes abnormal activation of ACVR1, leading to altered connective tissue and the conversion of muscle tissue to the secondary skeleton. This process eventually leads to the endothelial cells being transformed into mesenchymal stem cells and then into bone (Figure 8) [8].
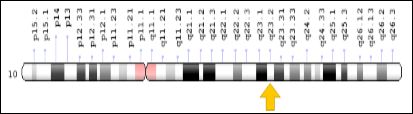
Figure 7: Schematic overview of chromosome 10 where the BMPR1A gene is located on the long arm of chromosome 10q23.2.

Figure 8: Schematic overview of chromosome 10 where the BMPR1A gene is located on the long arm of chromosome 10q23.2.
It should be noted that most cases of stone man syndrome (FOP) are caused by spontaneous mutations in gametes and since this syndrome follows an autosomal dominant inheritance pattern, so only one copy of the ACVR1 mutant gene for the emergence of FOP is sufficient. The mutated gene can be passed down from one parent to the next generation, but mostly because of the spontaneous mutation in the gametes. Also environmental epigenetic factors such as microbial bodies (viruses, bacteria, maternal nerve shock during zigzag cell dysfunction, malnutrition, poor lifestyle, bio-contaminated ecosystem and overuse and uncontrolled drug use) can induce gene mutation. ACVR1 have a role. However, the exact cause of the human stone syndrome why the mutation in the ACVR1 gene is spontaneous remains to be determined (Figures 9 and 10) [9].

Figure 9: Schematic overview of chromosome 10 where the BMPR1A gene is located on the long arm of chromosome 10q23.2.

Figure 10: Schematic overview of chromosome 14 where the BMP4 gene is located on the long arm of chromosome 14q22.2.
The Frequency of Stone Man Syndrome
Stone man syndrome (FOP) is a very rare genetic disorder of connective tissue that was first recognized in the 17th century. Of the approximately 3,000 people affected worldwide, only 800 have been officially recognized as human-stone syndrome. This disorder affects both men and women and all human relatives. The exact frequency of this syndrome is unclear, but since the disorder is very rare, the average incidence of the syndrome is 0.5 in 1 million or 1 in 2 million live births [10].
Diagnosis of Stone Man Syndrome
Diagnosis of stone man syndrome, or FOP, is usually mistaken, as some features of this disorder are common with other genetic syndromes. But it can be suspected by examining the toes of infants with FOP, who are usually large, with stone man syndrome. However, the most definitive test to confirm the diagnosis of stone man syndrome or FOP disease is molecular genetic testing for the ACVR1 gene to determine whether or not there is a mutation [11-14].
The Therapeutic Pathways of Stone Man Syndrome
Tissue biopsy should be avoided in this syndrome, as these tests may result in rapid bone formation in areas where the tissue is removed. Muscle infusions should also be avoided. In addition, patients with stone man syndrome should also avoid any falls, even simple falls, as this may cause blunt trauma (blunt damage) and promote abnormal bone growth. Viral diseases like influenza and influenza-like viruses, which may cause flares or abnormal bone growth, should also be seriously avoided. Patients who are susceptible to increased respira- tory infections should regularly use the appropriate antibiotic to prevent abnormal growth of the bones [15-18].
There is no effective and conclusive treatment for stone man syndrome (FOP). Certain types of medications are also used to relieve pain, the most important of which are corticosteroids and anti-inflammatory drugs. People with this syndrome may benefit from medical treatment. Special shoes, knee braces, braces, and other devices that help with weight and gait help people with stone man syndrome. Genetic counseling is important for families who want to have a healthy child, as well as for families with a family history of human stone syndrome [19].
History of Stone Man Syndrome
Stone man syndrome, or FOP, was first reported in the medical literature in the 17th century. FOP was called bone myositis at the time, and it was thought that muscle inflammation caused abnormal bone formation in patients. The disease was diagnosed in 1970 by Dr. Victor Almon McKusick (1921-2008), a physician and medical geneticist at Johns Hopkins University in Baltimore, Maryland, after discovering that soft tissue (such as ligaments) is in the process of disease. They have a major role renamed Progressive Bone Disease Fibrodysplasia (FOP). The best-known case of FOP is that of a man named Harry Eastlack (1933-1973) whose symptoms began at age 10 and six days before his 40th birthday, his body completely immobilized or immobilized. And was only able to move the lips. In November 1973, he died of pneumonia. In April 2006, an international team of researchers led by Dr. Eileen M. Shore, a cell and molecular biologist, and Dr. Frederick S. Kaplan, a research fellow at the University of Pennsylvania, published the results of their FOP research that revealed the mutation. In the gene ACVR1 has been reported as a genetic cause of FOP disease and has been reported as a human stone syndrome (Figure 11) [20,21].

Figure 11: Images of Mr. Harry Eastlack with stone man syndrome, who began his symptoms at age 10 and died at age 40.
Conclusion
Fibrodysplasia ossificans progressiva is a very rare and disabling disorder that, if misdiagnosed, can lead to unnecessary surgical intervention and disastrous results of early disability. We need to spread knowledge to physicians and patients’ family members about the disease, as well as its features for early diagnosis and how to prevent flareup of the disease to promote better quality of life in these patients.
References
- Asadi S, Jamali M, Bagheri R, Sadeh Dell S, Tohidirad M, et (2017) Book of Medical Genetics, Amidi Publications Iran.
- Kaplan FS, Le Merrer M, Glaser DL, Pignolo RJ, Shore EM, et al. (2011) Fibrodysplasia ossificans progressiva.
- Obamuyide HA, Ogunlade SO (2015) A Tumour for which Surgery will do more harm than good: A Case Report of Fibrodysplasia Ossificans Progres- Niger Postgrad Med J 22: 83-88.
- Saleh M, Commandeur J, Bocciardi R, Kinabo G, Hamel B (2015) Fibro- dysplasia ossificans progressiva with minor unilateral hallux anomaly in a sporadic case from Northern Tanzania with the common ACVR1c.617G>A Pan Afr Med J 22: 299.
- van Dinther M, Visser N, de Gorter DJJ, Doorn J, Goumans M-J, et (2010) ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. Journal of Bone and Mineral Research 25: 1208-1215.
- Shore Eileen M, Kaplan Frederick S (2008) Insights from a Rare Genetic Disorder of Extra-Skeletal Bone Formation, Fibrodysplasia Ossificans Pro- gressiva (FOP). Bone 43: 427-433.
- Genetics Home Reference, U.S. National Library of Medicine, August 2007. Accessed February 18, 2014.
- Martelli A, Santos AR Jr (2014) Cellular and morphological aspects of fibro- dysplasia ossificans Lessons of formation, repair, and bone bio- engineering. Organogenesis (Review) 10: 303-311.
- Kasper, DL, Fauci AS, Longo DL, et al. (2005) eds. Harrison’s Principles of Internal Medicine. 16th McGraw-Hill Companies. New York NY 2286.
- Kaplan FS, Shore EM (2003) Fibrodysplasia Ossificans Progressiva. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia PA 713-714.
- Chakkalakal S, Uchibe K, Convente M, Zhang D, Economides A, et (2016) Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice with the Human ACVR1 Fibrodysplasia Ossificans Pro- gessiva (FOP) Mutation. J Bone Miner Res 31: 1666-1675.
- Pignolo RJ , Bedford-Gay C, Liljesthröm M, Durbin-Johnson BP , Shore EM et al. (2016) The natural history of flare-ups in fibrodysplasia ossificans progressiva (FOP): A comprehensive global assessment. J Bone Miner Res 31: 650-656.
- Wang H, Lindborg C, Lounev V, Kim J, McCarrick-Walmsley R, et al. (2016) Cellular Hypoxia Promotes Heterotopic Ossification by Amplifying BMP Sig- naling. J Bone Miner Res 31: 1652-1665.
- Hatsell S, Idone V, Alessi Wolken D, Huany L, Kim H, et al.(2015) ACVR1 receptor mutation causes fibrodysplasai ossificans progressiva by imparting responsiveness to activing A. Sci Transl Med 7: 303ra137.
- Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, et (2012) An Acvr1 Knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res27: 1746-1756.
- Deirmengian GK, Hebela NM, O’Connell M, Glaser DL, Shore EM, et al. (2008) Proximal tibial osteochondromas in patients with fibrodysplasia ossifi- cans progressiva. J Bone Joint Surg Am 90: 366-374.
- Moore RE, Dormans JP, Drummond DS, Shore EM, Kaplan FS, et (2009) Chin-on-chest deformity in patients with fibrodysplasia ossificans progres- siva: a case series. J Bone Joint Surg Am 91: 1497-1502.
- Shen Q, Little SC, Xu M, Haupt J, Ast C, et al. (2009) The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo J Clin Invest 119: 3462- 3472.
- Lounev V, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment ADA, et al. (2009) Identification of progenitor cells that contribute to heterotopic J Bone Joint Surg Am 91: 652-663.
- Kitterman JA, Kantanie S, Rocke DM, Kaplan FS (2005) Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pedi- atrics 116: 654-661.
- Schaffer AA, Kaplan FS, Tracy MR, O’Brien ML, Dormans JP, et al. (2005) Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klip- pel-Feil syndrome: Clues from the BMP signaling pathway. Spine 30: 1379-
Citation: Asadi S, Aranian MR (2020) The Role of Genetics Mutations in Genes ACVR1, BMPR1A, BMPR1B, BMPR2, BMP4 in Stone Man Syndrome. J Hematol Hemother 5: 008.
Copyright: © 2020 Asadi S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and re- production in any medium, provided the original author and source are credited.
