*Corresponding Author:
Timothy Tramontana,
Department of Medical and Molecular Genetics, Indiana University School of Medicine, 975 West Walnut Street, Indianapolis, Indiana 46202-5251, USA
Tel: +1 2168443936
Email: ttramont@iu.edu
Abstract
Autosomal Recessive Cutis Laxa (ARCL) is a condition characterized by genetic heterogeneity and variable expressivity. In particular, compound heterozygous or homozygous mutations in the PYCR1 gene are the cause of both autosomal recessive cutis laxa IIB (ARCL2B) and type IIIB (ARCL3B), the latter of which is a subtype of de Barsy syndrome. In this article, we present an 8-month- old boy with progeroid features. Molecular testing subsequently identified novel compound heterozygous variants in the PYCR1 gene diagnostic of autosomal recessive cutis laxa based on clinical correlational evidence. This case illustrates the importance of recognizing the implications for prognosis, management and counseling in patients with novel compound heterozygous variants of the type found in our patient.
Keywords
Autosomal recessive cutis laxa; ARCL2B; ARCL3B; de Barsy syndrome; Progeroid; PYCR1
Introduction
PYCR1 encodes a mitochondrial enzyme called Pyrroline-5- Carboxylate Reductase 1 (P5CR1). This enzyme catalyzes the final step in proline biosynthesis. The gene’s highest expression is found in bone and skin, which are the tissues most affected in patients with Autosomal Recessive Cutis Laxa (ARCL) [1]. Although hypoprolinemia would be expected, with pathogenic mutations in PYCR1, serum proline levels in patients are generally within the normal ranges [1]. Therefore, other unidentified factors must have an impact on the serum proline levels. Both Autosomal Recessive Cutis Laxa IIB (ARCL2B, MIM: 612940) and type IIIB (ARCL3B,
MIM: 614438) are caused by pathogenic variants in PYCR1 and have phenotypic overlap. This overlap then creates a diagnostic dilemma for the clinician since the specific diagnosis can have prognostic implications. Both disorders are characterized by generalized cutis laxa, craniofacial abnormalities including, microcephaly, osteopenia, scoliosis, congenital hip dislocation, and psychomotor and growth delays. Additional features including progeroid appearance, strabismus, corneal opacities, hernias, short stature, hypotonia, athetoid movements, and severe intellectual disability. Pathogenic missense and splice-site variants of the PYCR1 gene are the most common reported mutations [2]. Interestingly, the most severe phenotypes are associated with missense variants in exons 4-6 [2] de Barsy syndrome is the more severe end of the phenotypic spectrum of ARCL3B.
Case Presentation
The male patient of African American and caucasian ethnicity was born to a 19-year-old G1P0 mother at 39 weeks gestation. His length was 45cm (3rd % tile), weight 2320g (2nd % tile) and occipitofrontal circumference was 32cm (6th % tile). The pregnancy was complicated by intrauterine growth restriction with an estimated fetal weight less than the 3rd % tile.
The family history was noncontributory. Delivery was via an uncomplicated C-section. Shortly after delivery, he became apneic, required positive pressure ventilation and was transferred to the Neonatal Intensive Care Unit (NICU). On exam, he was emaciated, was small for gestational age, had dolichocephaly, a tall forehead with bitemporal narrowing, mild orbital depression, shortened nasal bridge, small and dysplastic ears, adducted thumbs, arachnodactyly, reduced body fat associated with prominent superficial veins and hypotonia. Head and renal ultrasounds were normal. He was discharged from the NICU on day 12 of life.
At 8 months of age his progeroid facial features (Figure 1A and 1B) were quite striking and his skin was unusually wrinkled. He was still hypotonic with central hypotonia greater than peripheral. He also had developed contractures of all his fingers.
Results
A chromosomal microarray was completed and showed a variant of uncertain significance for a duplication in the SHOX gene (359.8 kb duplication at Xp22.33 or Yp11.32). A custom exome-based sequencing panel (GeneDx, Gaithersburg, MD) included 18 genes (ADAMTS2, ALDH18A1, ATP6VOA2, ATP6V1A, ATP6V1E1, B3GALT6, B4GALT7, BANF1, COL3A1, FBN1, IGF1R, KCNJ6, LMNA, PIK3R1, POLD1, PYCR1, WRN, ZMPSTE24) associated with progeria, conditions with progeria-like features, lipodystrophies, and connective tissue disorders, which can have progeroid features.
This latter study revealed compound heterozygous missense variants involving the PYCR1 gene (NM_006907.2) (Figure 2A). The first mutation was denoted as: c.356G>A (p.Arg119His) and classified as pathogenic. The second was: c.355C>T (p.Arg119Cys), and was classified as likely pathogenic. Both missense alterations were located inexon 4. The Arg119His variant has been reported previously in a compound heterozygous and homozygous state in unrelated individuals with ARCL. The Arg119Cys variant has been previously reported in the homozygous state in a patient with De Barsy syndrome.
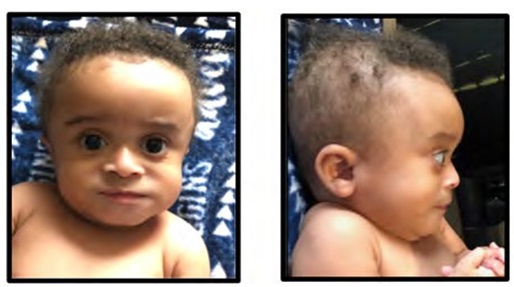
Figure 1: Patient viewed at age 8 months. Note patchy hair growth and laxity of skin with prominence along the dorsum of both hands and feet. A Facial view. Note the depressed nasal bridge, pinched nose appearance and tall, broad forehead; B) Lateral facial view. Observe the dolichocephaly and bitemporal alopecia; Hands and legs/feet.
Note: The significant wrinkling of skin on the dorsum of hands, legs and feet.
Discussion
Here we present a patient with PYCR1-related cutis laxa caused by compound heterozygous pathogenic mutations in PYCR1. Cutis laxa is a disorder, which can be inherited in an autosomal dominant, X-linked or autosomal recessive pattern [2]. Among the Autosomal Recessive Cutis Laxa (ARCL) syndromes, are the PYCR1-related cutis laxa disorders. These disorders share a more severe clinical phenotype have considerable variable phenotype and are genetically heterogeneousde Barsy syndrome is at the severe end of the spectrum. Both ARCL2B and ARCL3B (de Barsy syndrome) have phenotypic overlap, but differ in prognoses.
Facial dysmorphisms include thin and narrow nose, prominent forehead and progeroid-like appearance, while the neurologic features are hypotonia, developmental delay, varying levels of intellectual disability, and dysgenesis or agenesis of the corpus callosum. Other features include cutis laxa, congenital hip dislocation, joint laxity, osteopenia and osteoporosis, adducted thumbs, and varying degrees of lipodystrophy with visible subcutaneous vessels on the chest. Though not seen in all patients with ARCL2B, wormian bones, athetoid movements, lipodystrophy and cataracts are highly specific for ARCL2B [3]. Typical features of ARCL3B include agenesis of the corpus callosum, corneal clouding, hypotonia, progeroid appearance and short stature, movement disorder and profound intellectual disability. Additional phenotypic features including agenesis of the corpus callosum, cataracts, corneal clouding, athetoid movements and contractures make the diagnoses more likely. Further, the neonatal period is characterized by frontal bossing, thin nose with hypoplastic alae nasi and deep set eyes. As the patient matures, an elongated and triangular face with prominent mandible and strabismus can develop. Our patient has features of ARCL3B that include IUGR, progeroid features, lax skin, triangulated face, bilateral thumb contractures and arachnodactyly, athetoid movements of the upper extremities, mild upper extremity hypertonicity and truncal hypotonia.
Patients with PYCR1-related cutis laxa conditions can also have blue sclerae and inguinal hernias, which have been reported in approximately 25 and 50% of published cases, respectively [2]. Our patient had both of these findings. Visual abnormalities in this condition can include cataracts and corneal clouding. Our proband does not appear to have these latter findings, but has yet to have an ophthalmologic exam. Although hearing abnormalities are not part of the phenotypic spectrum, our patient did fail his hearing screen after birth and audiologic reevaluation was PYCR1-related cutis laxa has been well characterized molecularly. The Arg119H is variant has been reported previously in both compound heterozygous and homozygous states in unrelated individuals with ARCL [4]. Functional studies have demonstrated that the two site mutations, Arg119Gly and Arg119H is, could influence the function of the P5CR1 protein, which is likely the pathomechanism for ARCL [5].
The R119C variant in PYCR1 has previously been reported in the homozygous state in a patient with de Barsy syndrome [1]. Non- conserved amino acid substitutions, in silico analysis, and clinical correlation have supported a deleterious effect of this amino acid change. Comparison with six vertebrate PYCR1 orthologs revealed that the R119 residue is highly conserved (Figure 2B). All the variants that have been reported previously were predicted to be pathogenic (Figure 2C). Residue R119, along with other key residues (likely most functionally relevant being Arg251, Arg266 and Val258) that form part of the lateral surface of the PYCR1 monomer. These residues could make contact to the neighbouring PYCR1 homodimer and be involved in homodecamer formation [2]. As a result, this can interfere with overall protein stabilization thus protein expression.
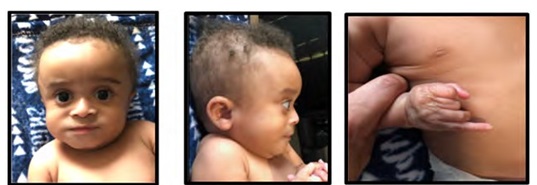
Figure 2: Molecular data. (A) Family pedigree and segregation of the two variants c.355C>T (p.R119C) and c.356G>A (p.R119H). (B) Schematic representation of the genomic structure of PYCR gene. Exons are depicted to scale as blue rectangles and the horizontal bars represent introns. Previous published variants are shown below and above the exons. The two variants detected in this study are shown in red. C) Clustal W multiple alignment analysis shows highlevel evolutionary conservation of the human R119 residue in PYCR1 across multiple species (boxed in red).
One of the significant features of the patient we present here is that he has a compound heterozygous genotype not encountered before. Whether or not his phenotype will be similar to ARCL2B or the more severe ARCL3B end of the spectrum remains to be seen. This type of situation obviously creates a prognostic dilemma for the provider as parents question the clinical course of their children. In our case, we explained that PYCR1-related autosomal recessive cutis laxa conditions represent a spectrum that essentially declares itself over time with development of additional features. A key element in monitoring patients with these disorders is the development of severe intellectual impairment phenotype, which would be consistent with a diagnosis of ARCL3B (de Barsy syndrome).
Conclusion
The case presented here demonstrates a few key points regarding PYCR1- related ARCL disorders. First, there is a wide differential for a progeroid presentation including connective tissue disorders like neonatal Marfan syndrome, Hutchinson-Gilford progeria syndrome and a host of cutis laxa related disorders. Secondly, as in this case, the identification of a pathogenic variant, although helpful in establishing or confirming the diagnosis in most situation, can still present the provider with a clinical dilemma. This clinical dilemma has direct implications for prognostic prediction and guidance in counseling families on short- and long-term risks with regard to their child’s specific condition. With progeroid-like cases, both ARCL2B and ARCL3B (aka de Barsy syndrome) must be considered. Third and likely most important is the phenotypic overlap between ARCL2B/3B and that ultimately the presentation is more severe for ARCL3B, particularly the severe intellectual disability. Finding a pathogenic mutation in PYCR1 does not tell one the prognosis for the affected individual. The specific type of ARCL2B/3B will depend on the course of the patient.
Acknowledgments
The authors would like to thank this family for allowing us to participate in the care and discussion of this patient..
Conflicts of Interest
Funding
Indiana University School of Medicine has provided the salaries of the authors..
References
- 1. Reversade B, Escande-Beillard N, Dimopoulou A, Björn Fischer B, Chng SC, et al. (2009) Mutations in PYCR1 cause cutis laxa with progeroid Nat Genet 41: 1016-1021.
- 2. Dimopoulou A, Fischer B, Gardeitchik T, Schröter P, Kayserili H, et al. (2013) Genotype–phenotype spectrum of PYCR1-related autosomal re- cessive cutis Mol Genet Metab 110: 352-361.
- 3. Kariminejad A, Afroozan F, Bozorgmehr B, Ghanadan A, Akbaroghli S, et al. (2017) Discriminative features in three autosomal recessive cutis laxa syndromes: Cutis laxa IIA, cutis laxa IIB, and geroderma osteoplas- International Journal of Molecular Sciences 18: 635.
- 4. Scherrer DZ, Baptista MB, Matos AH, Maurer-Morelli CV, Steiner CE (2013) Mutations in PYCR1 gene in three families with autosomal re- cessive cutis laxa, type 2. European Journal of Medical Genetics 56: 336-339.
- 5. Li L, Ye Y, Sang P, Yin Y, Hu W, et al. (2017) Effect of R119G Mutation on Human P5CR1 Dynamic Property and Enzymatic Activity. Biomed Res 4184106.
Citation:Tramontana T, Vetrini F, Weaver DD, Conboy E, Helm B, et al. (2021) Novel Combination of Compound Heterozygous PYCR1 Gene Variants in Autosomal Recessive Cutis Laxa: Implications for Prognosis, Management and Counseling. J Case Repo Imag 5: 037.
Copyright: © 2021 Tramontana T, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
