*Corresponding Author:
Jayesh J Sheth,
Department of Biochemical and Molecular Genetics, Institute of Human Genetics, Gujarat, India
Tel: +91 7926961414
Email: jshethad1@gmail.com
Abstract
Pediatric DIP is known to occur due to defects in surfactant pro- tein production genes, however, its association with lysosomal stor- age disorders, is less known and understudied. Here we report a couple with two earlier children who died with respiratory distress at 5 years and 8 months, respectively. Clinical exome sequencing study of the couple was reported with heterozygous pathogenic nonsense variant c.141C>A (p. Cys47Ter) in the NPC 2 gene. Sub- sequent pregnancy was diagnosed with an affected foetus carrying aforementioned homozygous variant. This is the first report of NPC2 masked with clinical presentation of DIP.
Keywords
Interstitial pneumonitis; Niemann-Pick disease
Introduction
Niemann Pick-C type 2 (NPC2) usually present in early infancy with organomegaly as the major phenotype attributed to an accumulation of lipid laden macrophages [commonly termed as Niemann-Pick cells (NP cells)] in the liver, spleen, bone marrow, central nervous system and also the lungs [1-3]. Though several studies have been reported with mainly respiratory signs like interstitial pneumonia, overloaded alveolar macrophages in alveolar spaces and walls in a baby affected with NPC [1,2,4,5]. Here with we describe a case with severe pulmonary involvement in two children died in childhood suspected with surfactant protein deficiency and during third pregnancy detailed genomic study of the family has revealed the cause due to NPC2 gene mutation
Case Report
A non-consanguineous couple was referred for genetic counseling during third gravida since their previous two children died due to respiratory distress at the age of 5 years and 8 months, respectively.
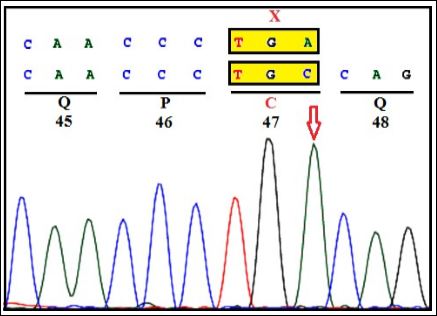
Figure 1A: CVS: Homozygous c.141C>A (C47X) in exon-2 of NPC2 gene.
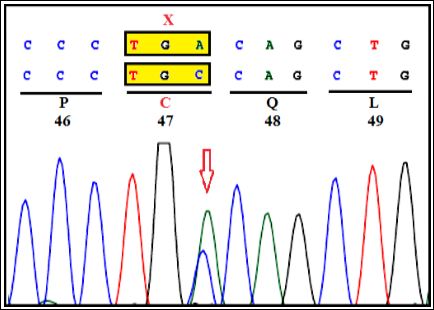
Figure 1B: Mother: Heterozygous c.141C>A (C47X) in exon-2 of NPC2 gene.

Figure 1C: Father: Heterozygous c.141C>A (C47X) in exon-2 of NPC2 gene.

Table 1: Variant identified by clinical exome sequencing in a family with DIP.
Note: AR: Autosomal Recessive; Het: Heterozygous; Hom: Homozygous; NR: Not Reported; NGS: Next Generation Sequencing
The first full term child was born with normal delivery and a birth weight of 3.49 kg. Respiratory difficulty was observed at 3 months which progressed to severe cough and tachypnea. The child was subjected to intensive care for bilateral bronchopneumonia without any response to treatment. X-ray chest showed bilateral persistent consolidation. Though chest CT image of the child was not available with the family as has been deposited in the hospital record. A water-soluble contrast swallow test ruled out trachea-oesophageal fistula but gastrographine study showed gastro-esophageal reflux. He continued to have tachypnea even after anti-reflux formula. Investigations including an immunoglobulin test, barium meal study and bronchoscopy were normal. Echocardiogram ruled out heart anomaly. He was on anti-tuberculosis therapy for nine months.
Four months later, he was presented with breathing difficulty and cyanosis. He was severely distressed and not responding to aggressive treatment with nebulized salbutamol and ipratropium bromide in addition to intravenous hydrocortisone and aminophylline. He was on ventilation for four days followed by a ten-day course of intravenous antibiotics.
At the age of three years an interstitial lung disease was suspected. Lung biopsy of the first child was performed which has shown plump macrophages filling often distending all alveoli. The cells showed abundant granular cytoplasm and small oval based nuclei. In some areas alveolar septa were thickened due to significant lymphocyte infiltrate. In others, the alveolar septa were of normal thickness or even thinner than normal. No traces of necrosis, hyaline membranes and fibrin deposition were observed in alveoli leading to histological diagnosis of Desquamative Interstitial Pneumonitis (DIP). Since family approached us after three years to know the cause, no paraffin blocks or slides were available with them except a copy of histopathology report. His health progressively deteriorated by 4 years and the child finally succumbed to death by 5 years. The second female sibling also had similar symptoms and died at 8 months without any detailed investigations except she was kept on ventilator due to severe respiratory distress.
A genetic approach to the diagnosis of CHILD (CHILDREN INTERSTITIAL LUNG DISEASE) has been reviewed by Nogee LM in the elder child [6]. He recommended that the newborns with respiratory distress syndrome and persistent pulmonary hypertension should be evaluated for ABCA3, SFTPC and SFTPB mutations [6]. Since alteration in ABCA3 gene results in fatal surfactant protein deficiency leading to early death. The mother under report (in absence of affected live children) was initially investigated for ABCA3 gene defect (both the coding exons and flanking intron-exon boundaries) through bidirectional Sanger sequencing on ABI 3500. No sequence variation/s was identified that could be associated to lung disease. Further, sequence analysis of SFTPC gene (encoding surfactant protein C) and SFTPB gene (encoding surfactant protein B) from both the parents was performed by bidirectional Sanger sequencing on ABI3500. No variant/s in either of the gene were detected that may explain the cause of surfactant protein deficiency in the child.
During third gravida, extensive genetic investigations were carried out. Clinical exome study of the parents was carried by selective gene capture using custom capture kit. The libraries were sequenced to 80 to 100X coverage on Illumina sequencing platform. Our study has identified heterozygous nonsense variant c.141C>A (p. Cys47Ter) of the NPC2 gene (OMIM*601015; NP_006423.1; NM_006432.4) (Figure 1A) and bidirectional Sanger sequencing on Seq studio further confirmed the variant in both parents (Figure 1B). Based on this analysis, prenatal diagnosis was conducted on chorionic villi in third gravida at 12 weeks. A homozygous mutation in exon-2 of NPC2 gene (c.141C>A) was detected (Figure 1C). Thus, trio study has confirmed the diagnosis as NPC2 in the third gravida fetus. The parents opted for termination of pregnancy. The identified variant is reported as ‘pathogenic’ in ClinVar database (RCV000020644) that results in protein truncation (Table 1).
Discussion
The mutation c.141C>A (p.Cys47Ter), identified in present case was previously observed in a child with alveolar proteinosis and is now observed to be associated with another interstitial lung condition, namely DIP masking NPC further suggesting a likely common hot spot mutation in children with NPC2 and pulmonary involvement. Several groups have documented lethal and pronounced lung involvement in NPC2 [1,2,5].
The case under study provides evidence for a novel possible etiology of DIP due to NPC-2 mutation. The gene located at 14q24.3 encodes NPC2 protein [4] which works in conjunction with NPC1 to facilitate the transfer of cholesterol to other cell organelles that are responsible for maintaining intracellular cholesterol homeostasis. NPC2, localized in the lumen of lysosome binds to free cholesterol and initiate a hydrophobic exchange of cholesterol with NPC1 protein. This is further involved in facilitating cholesterol transport to the golgi apparatus, that further passes on to the plasma membrane or the endoplasmic reticulum by an unknown mechanism [8].
Therefore, NPC2 mutation likely results into a non-functional protein and subsequent perturbed cellular cholesterol homeostasis. This was demonstrated in a study in which cells transfected with mutant, nonfunctional NPC genes exhibited cholesterol accumulation [9-11]. This indicates that mutations in the NPC genes may lead to lipid storage dysregulation in lysosomes.
This report highlights the presentation of variable pulmonary manifestations with NPC2, draws attention to heterogeneity of phenotypes, and accentuates the need for investigating NPC2 gene in cases reported with histology of DIP without splenomegaly in the young children.
Acknowledgments
We thank Dr. Harsh Sheth and Dr. Sunil Trivedi for their expert advice and suggestions. We express gratitude to the patients’ parents for their cooperation.
References
- Guillemot N, Troadec C, de Villemeur TB, Clément A, Fauroux B (2007) Lung disease in niemann-pick disease. Pediatr Pulmonol 42: 1207-1214.
- Sheth J, Joseph JJ, Shah K, Muranjan M, Mistri M, et al. (2017) Pulmonary manifestations in Niemann-Pick type C disease with mutations in NPC2 gene: Case report and review of literature. BMC Med Genet 18: 5.
- Vanier MT (2010) Niemann-Pick disease type Orphanet J Rare Dis 5: 16.
- Pin I, Pradines S, Pincemaille O, Frappat P, Brambilla E, et al. (1990) A fatal respiratory form of type C Niemann-Pick Arch Fr Pediatr 47: 373-375.
- Schofer O, Mischo B, Püschel W, Harzer K, Vanier MT (1998) Early-lethal pulmonary form of Niemann-Pick type C disease belonging to a second, rare genetic complementation group. Eur J Pediatr 157: 45-49.
- Nogee LM (2010) Genetic basis of children’s interstitial lung disease. Pedi- atr Allergy Immunol Pulmonol 23: 15-24.
- Chikh K, Rodriguez C, Vey S, Vanier MT, Millat G (2005) Niemann-Pick type C disease: Subcellular location and functional characterization of NPC2 proteins with naturally occurring missense mutations. Hum Mutat 26: 20-20.
- Garver WS, Heidenreich RA (2002) The Niemann-Pick C proteins and trafficking of cholesterol through the late endosomal/lysosomal system. Curr Mol Med 2: 485-505.
- Liebow AA, Steer A, Billingsley JG (1965) Desquamative interstitial pneu- Am J Med 39: 369-404.
- Tubbs RR, Benjamin SP, Reich NE, McCormack LJ, Van Ordstrand HS (1977) Desquamative interstitial pneumonitis. Cellular phase of fibrosing Chest 72: 159-165.
- Sugimoto Y, Ninomiya H, Ohsaki Y, Higaki K, Davies JP, et (2001) Ac- cumulation of cholera toxin and GM1 ganglioside in the early endosome of Niemann-Pick C1-deficient cells. Proc Natl Acad Sci 98: 12391-12396.
Citation:Sheth JJ, Bhavsar R, Sheth FJ (2020) Niemann-Pick Disease Type C2 Pre- sented as Desquamative Interstitial Pneumonitis in a Non-Consanguineous Family. J Perina Ped 4: 010.
Copyright: © 2020 Sheth JJ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
