*Corresponding Author:
Jinjun Cheng,
Department of Pathology, Molecular and Cell-Based Medicine, USA.
Tel: 212-241-1440
E-mail: Jinjun.cheng@mountsinai.org
Abstract
First described in 1972, Kikuchi-Fujimoto Disease (KFD) is a rare benign syndrome of necrotizing lymphadenopathy and most commonly affects young females. Patients usually present with cervical lymphadenopathy which can be painful, some patients have adenopathy involving multiple regions and fever. Histologically, the early proliferative phase of KFD is characterized by presence of many immunoblasts with prominent nucleoli, resembling Diffuse Large B-Cell Lymphoma (DLBCL). With the presence of necrosis and many cytotoxic T cells with cytological atypia, KFD can also mimic T-cell lymphoma in histology. Therefore, it can be challenging to differentiate KFD from lymphoma both clinically and morphologically. Distinction between KFD and Primary Mediastinal B-Cell Lymphoma (PMBL), especially in the absence of mediastinal mass, has not been reported. Here we present a unique case showing some morphological overlapping features between a PMBL and KFD.
Keywords
Differential diagnosis; Kikuchi Fujimoto Disease; Primary Mediastinal B-cell Lymphoma
Background
First described in 1972, Kikuchi-Fujimoto Disease (KFD) is a rare benign syndrome of necrotizing lymphadenopathy and mostly affects young females [1,2]. Patients usually present with cervical lymphadenopathy which can be painful, some patients have adenopathy involving multiple regions and fever [3]. The underlying etiology is uncertain but viral infection and autoimmune pathogenesis have been proposed [4,5]. Radiographically, KFD exhibits increased Standardized Uptake Value (SUV) on PET/CT scan [6]. Histologically, the disorder shows three phases including proliferative, necrotic, and resolution phase [3-5,7-9]. The early proliferative phase is characterized by presence of many immunoblasts with prominent nucleoli, resembling Diffuse Large B-Cell Lymphoma (DLBCL). With the presence of necrosis and many cytotoxic T cells with cytological atypia, KFD can also mimic T-cell lymphoma in histology. Therefore, it can be challenging to differentiate KFD from lymphoma both clinically and morphologically. Distinction between KFD and Primary Mediastinal B-Cell Lymphoma (PMBL), especially in the absence of mediastinal mass, has not been reported.
Case Report
We present a case of a young malewith no significant past medical history who presented with waxing and waning cervical lymphadenopathy with occasional fever, mild night sweat, and 5 pounds weight loss over one month. The initial cytological evaluation of right cervical lymph node showed polymorphous lymphocytes with increased histiocytes (Figure 1A). Excisional biopsy of the lymph nodes was then performed, which contained a focal area enriched with histiocytes and mild necrosis and characterized by a conspicuous absence of granulocytes. Some histiocytoid cells showed twisted/crescentic nuclei (Figures 1B and D). CD68 highlighted numerous histiocytes in some areas. CD123 stained rare plasmacytoid dendritic cells (data not shown). However, away from the histiocyte enriched areas; there were abundant immunoblasts and centroblastlike atypical large lymphocytes (Figure 1C and E). These findings resembled necrotic and proliferation phases of KFD, but also raised a concern for a B-cell lymphoma.
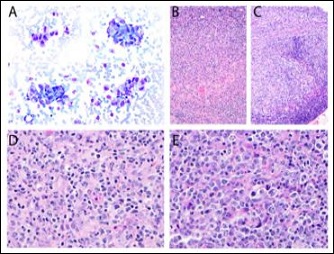
Figure 1: Right cervical lymph nodes. Increased clusters of histiocytes are present on the cytological evaluation (A, 400X). The excision biopsy contains areas of many histiocytes and focal necrosis (B, 100X; D, 400X), and some other areas with abundant immunoblasts and centroblast-like atypical lymphocytes (C, 100X; E, 400X). These patterns resemble different phases of KFD on H&E sections.
Fine needle aspiration of the left cervical lymph node showed few atypical large lymphocytes in a background of polymorphous lymphocytes (Figure 2A). The subsequent excision biopsy showed areas with confluent sheets of atypical large lymphocytes exhibiting similar cytomorphology to the ones seen in cytology specimen (Figure 2B). These cells were positive for CD20, CD23, CD30, MYC (in a subset), BCL2, BCL6, and high Ki67 proliferation rate (Figures 2C-I). EBER was negative. Concurrent flow cytometry detected atypical B-cell population negative for surface light chain expression (Figure 2J). This case was further reviewed with outside expert hematopathologists and the final diagnosis was B-cell lymphoma, most consistent with PMBL.
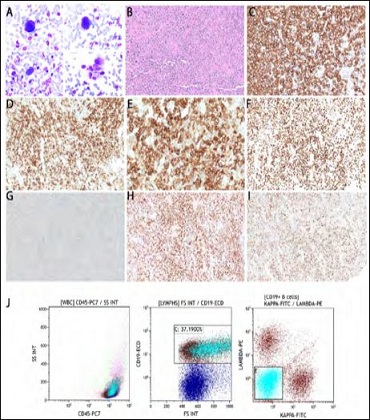
Figure 2: Left cervical lymph nodes. Numerous atypical lymphocytes are present on the cytological evaluation (A, 400X) and excisional biopsy (B, 100X), with the full immune phenotype of CD20+ (C), CD23+ (D), CD30+ (E), Ki67 90% (F), MYC weakly+ in a subset (G), BCL2 (H), and BCL6(I, 100X). Concurrent flow cytometry detected atypical B-cell population with negative surface light chain and high forward side scatter (aqua population, J).
Next Generation Sequencing of 154 gene panel detected the alterations noted on PD-L1, JAK2, PD-L2 and CIITA. CD274 (PD-L1) amplification, JAK2 amplification, PDCD1LG2 (PD-L2) amplification, CIITA splice site 52_52+31del32 and other somatic mutations including ARID1A N15fs*91, B2M M1I, CD58 S87fs*2, S158fs*10, PHF6 C295fs*72, SOCS1 F101fs*17, A35fs*50, L150*, TNFAIP3 splice site 986+2T>A, R713fs*8, TP53 R273C, XPO1 E571K.
During workup, PET/CT scan showed multiple intensely FDG avid lymph nodes in the neck, chest, and abdomen without a large mediastinal mass. The patient started treatment with dose adjusted EPOCH-R regimen (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin and rituximab).
Discussion
KFD was first described by Kikuchi and Fujimoto in Japan in 1972 as histiocytic necrotizing lymphadenitis, a self-limiting syndrome with distinct histological features [2]. The diagnosis is based primarily on the lymph node biopsy in the setting of typical clinical symptoms. Our patient has the overlapping symptoms with KFD [5,7-9]. The histologic features of KFD include three phases. During the early proliferative phase, the biopsy often shows patchy areas with a proliferation of medium-to-large lymphoid cells, histiocytes, and plasmacytoid dendritic cells [5]. Neutrophils, eosinophils, and plasma cells are rare. At necrotic phase, there is a focal to extensive necrosis with admixed karyorrhectic debris and absence of mature neutrophils, which usually is circumscribed by medium-to-large sized lymphoid cells and crescent shaped histiocytes. The last resolution phase mainly shows presence of many foamy macrophages [5,7]. Our patient’s biopsy resembles proliferative and focally necrotizing patterns of KFD. However, although rare genetic abnormalities are reported in KFD [10], numerous chromosomal and somatic mutations in our patient argue against KFD, and the differential diagnoses focus on diffuse large B-cell lymphoma and Primary Mediastinal Large B-Cell Lymphoma (PMBL).
PMBLis a separate but rare subtype of DLBCL in the WHO classification, which arises in the thymus and usually affects young female adults [9]. Although majority of patients present with an enlarging mediastinal mass that infiltrates nearby structures and causes symptoms of compression, rare PMBL may present as lymphadenopathy without mediastinal mass [9,11].
Morphologically, the tumor cells of PMBL show variation in cell and nuclear size, and resemble centroblasts, immunoblasts, anaplastic or Hodgkin Reed Sternberg cells [9,12,13]. Background fibrosis and sclerosing are common [9,14]. Phenotypically, PMBL retains B-lineage markers such asCD19, CD20, PAX5, while absence of surface and cytoplasmic immunoglobulin is relatively common [11,13]. CD30 expression is usually homogenous [9,11,13]. Genetically, PMBL shows clonal rearrangement of immunoglobulin heavy and/or light chains. Chromosomal rearrangement of BCL6, BCL2, and MYC are rare, with overall less than 5% of cases [9,13,14]. The most frequent chromosomal abnormalities include gains or amplifications of chr9p, containing genes of JAK2, PDL1 and PDL2 [9,14,15]. Activation mutations of JAK-STAT and NFκB pathways are seen in high percentage of diseases [9,11,12,14-17]. Our case demonstrated classical chr9p gains and somatic mutations involving JAK-STAT and NFκB pathways. The cytologicaland histological differential diagnoses of PMBL include DLBCL, NOS, classic Hodgkin’s disease, and other entities. However, in our case, PMBL is favored based on morphology, flow cytometry, and molecular features.
Acknowledgement
We appreciate Dr Elaine Jaffe at National Institute of Health and hematopathologist at Memorial Sloan Kettering for a consult opinion on this case.
References
- Dorfman RF, Berry GJ (1988) Kikuchi’s histiocytic necrotizing lymphadenitis: An analysis of 108 cases with emphasis on differential Semin Diagn Pathol 5: 329-345.
- Shirakusa T, Eimoto T, Kikuchi M (1988) Histiocytic necrotizing lymphad Postgrad Med J 64:107-109.
- Perry AM, Choi SM (2018) Kikuchi-Fujimoto Disease: A Review. Arch Pathol Lab Med 142: 1341-1346.
- Hollingsworth HC, Peiper SC, Weiss LM, Raffeld M, Jaffe ES (1994) An investigation of the viral pathogenesis of Kikuchi-Fujimoto Lack of evidence for Epstein-Barr virus or human herpesvirus type 6 as the causative agents. Arch Pathol Lab Med 118: 134-140.
- Scott GD, Kumar J, Oak JS, Boyd SD, Raess PW, et al. (2020) Histology-Independent Signature Distinguishes Kikuchi-Fujimoto Disease/ Systemic Lupus Erythematosus-Associated Lymphadenitis From Benign and Malignant Am J ClinPathol 154: 215-224.
- Tsujikawa T, Tsuchida T, Imamura Y, Kobayashi M, Asahi S, et (2011) Kikuchi-Fujimoto disease: PET/CT assessment of a rare cause of cervical lymphadenopathy. Clin Nucl Med 36: 661-664.
- Sukswai N, Jung HR, Amr SS, Ng SB, Sheikh SS, et (2020) Immunopathology of Kikuchi-Fujimoto disease: A reappraisal using novel immunohistochemistry markers. Histopathology 77: 262-274.
- Willemze R, Cerroni L,Kempf W, Berti E, Facchetti F, et al. (2019) The 2018 update of the WHO-EORTC classification for primary cutaneous Blood 133: 1703-1714.
- Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, et (2016) The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127: 2375-2390.
- Robertson KE, Forsyth PD, Batstone PJ, Levison DA, Goodlad JR (2007) Kikuchi’s disease displaying a t(2:16) chromosomal transloca J Clin Pathol 60: 433-435.
- Yuan J, Wright G, Rosenwald A, Steidl C, Gascoyne RD, et al. (2015) Identification of Primary Mediastinal Large B-cell Lymphoma at Nonmediastinal Sites by Gene Expression Profiling. Am J Surg Pathol 39: 1322-1330.
- Hoda RS, Picklesimer L, Green KM, Self S (2005) Fine-needle aspiration of a primary mediastinal large B-cell lymphoma: A case report with cytologic, histologic, and flow cytometric Diagn Cytopathol 32: 370-373.
- Oschlies I, Burkhardt B, Salaverria I, Rosenwald A, d’Amore ES, et al. (2011) Clinical, pathological and genetic features of primary mediastinal large B-cell lymphomas and mediastinal gray zone lymphomas in chil Haematologica 96: 262-268.
- Van Roosbroeck K, Ferreiro JF, Tousseyn T, van der Krogt JA, Michaux L, et (2016) Genomic alterations of the JAK2 and PDL loci occur in a broad spectrum of lymphoid malignancies. Genes Chromosomes Cancer 55: 428-441.
- Wang Y, Wenzl K, Manske MK, Asmann YW, Sarangi V, et (2019) Amplification of 9p24.1 in diffuse large B-cell lymphoma identifies a unique subset of cases that resemble primary mediastinal large B-cell lymphoma. Blood Cancer J 9: 73.
- Mottok A, Wright G, Rosenwald A, Ott G, Ramsower C, et (2018) Molecular classification of primary mediastinal large B-cell lymphoma using routinely available tissue specimens. Blood 132: 2401-2405.
- Pittaluga S, Nicolae A, Wright GW, Melani C, Roschewski M, et al. (2020) Gene Expression Profiling of Mediastinal Gray Zone Lymphoma and Its Relationship to Primary Mediastinal B-cell Lymphoma and Classical Hodgkin Lymphoma. Blood Cancer Discov 1: 155-161.
Citation: Cheng J, Lu D, Carmel A, Kalac M, Teruya-Feldstein J (2021) An Unusual Case of Primary Mediastinal B-cell Lymphoma Resembling Kikuchi Fujimoto Disease. J Tissue Biol Cytol 4: 008.
Copyright: © 2021 Cheng J, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
